

More on that later.
The latest incarnation of a priority review track is the Breakthrough Devices Program, the spiritual successor to the Expedited Access Pathway and Innovation Pathway.
For most companies, it means that a new medical device could be brought to market more quickly. However, it’s not necessarily that simple.
New data from JAMA Internal Medicine suggests that Class III products are more likely to have quality issues – and ultimately, even be subject to longer review time than those that endure the standard review process. But before we examine the issues, let’s take a look at:
- What the program is
- How eligibility is determined
What is it?
The breakthrough devices program is a voluntary program for specific medical devices (or combination products with pharma components) that provide more effective treatment or diagnoses of serious medical conditions. The goal is simple – to ensure that medical devices having significant patient benefit in the short term can be brought to market more quickly through premarket approval application (PMA), 510(k) clearance, and Devo Marketing clearance.
Is my product eligible?
The FDA lays out a few guidelines so that medical device companies can self-dicern eligibility prior to submission:

The Problems
Let’s look at the data. Overall, the number of devices granted breakthrough eligibility has increased increased 809% from 2016 to 2019, and it’s not looking to slowdown. Quality data from JAMA shows why the FDA needs to balance the patient benefit of breakthrough technology and safety.
Researchers at New York University and the University of California recorded how long the FDA took to approve Class III devices from 2005 to 2015. Furthermore, they looked into the safety records of those products1.
The comparison was staggering:
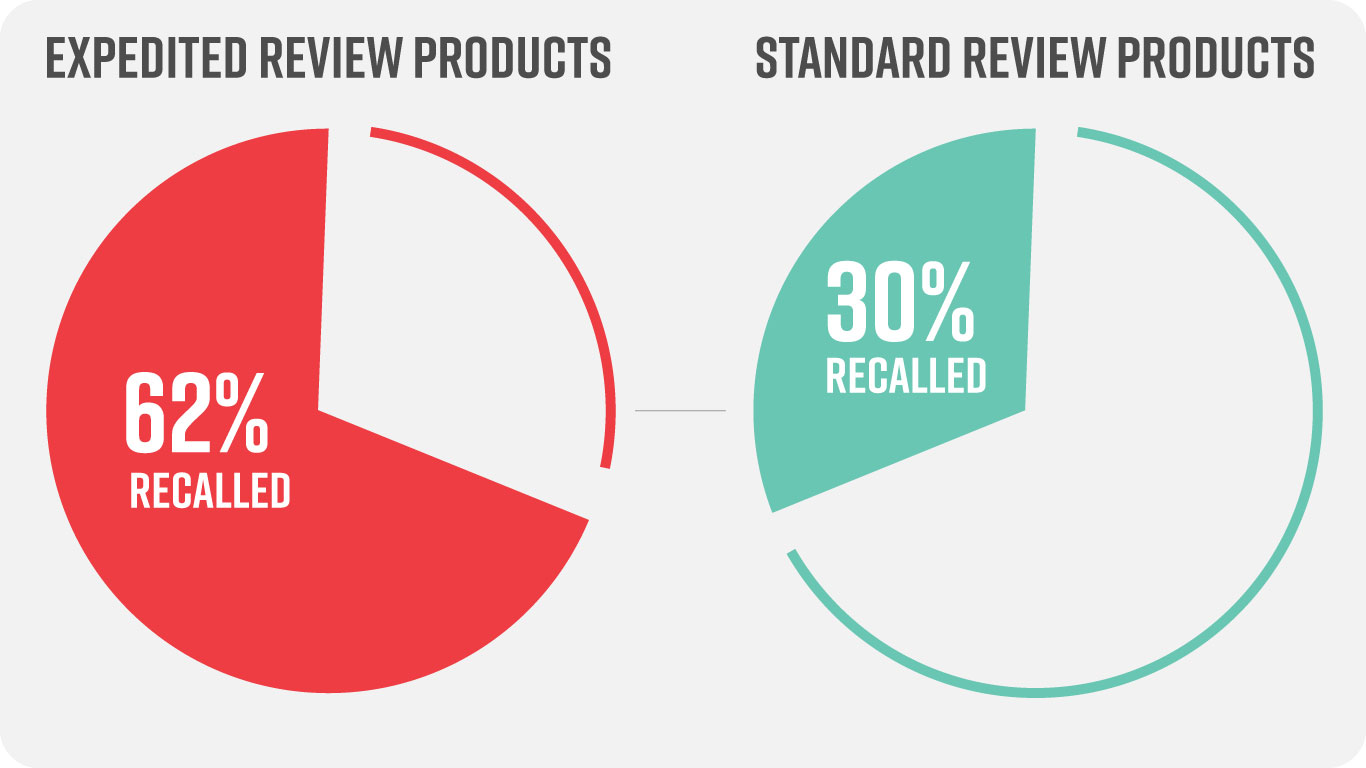
Additional, expedited products are more likely to have longer review cycles:

There are logical reasons for time and quality difference:
- New, novel technology is not understood
- The technology is groundbreaking and hasn’t been tested on large populations
- The devices are generally more complicated than products in standard review
- The products in expedited review represent a smaller sample size
Despite the issues, there is a silver lining – devices receiving priority review are not associated with more negative patient outcomes overall.
For more information, we recommend reading the entire JAMA report here.